Variant calling with bcftools
First let's see how to use a simple pipeline to identify genetic variants using bcftools mpileup and bcftools call.
As this suggests the process has two steps. In the first step (the mpileup step), we process the reads, identify likely alleles, and compute genotype likelihoods. In the second calling step these are used to jointly call variants and genotypes across the samples.
Let's get started. Move to a terminal window and make sure you are in the folder
variant_calling_and_imputation:
cd ~/variant_calling_and_imputation
As you should see, there are a number of aligned read files (BAM files) from the GWD population
("Gambians from the Western Division") in the reads/ folder. We have subsetted these down so they
cover the FUT2 gene (which we looked at briefly in the challenge question this morning).
Let's use bcftools to compute genotype likelihoods:
bcftools mpileup -Oz -f GRCh38_chr19.fa.gz -o GWD_FUT2_pileup.vcf.gz reads/*.bam
Note. This will take a minute or so to run, as the command reads all the alignment files and the FASTA reference sequence, and inspects the pileup much like we did manually this morning.
Let's go straight ahead and implement calling:
bcftools call -m -v -Oz -o GWD_FUT2_calls.vcf.gz GWD_FUT2_pileup.vcf.gz
Note. The -m option means 'use the newer multi-allele calling algorithm', and -v means
'only output variant sites'. In both commands we wrote -Oz to mean 'output compressed VCF format'.
Inspecting the pileup and calls output
Both the pileup and call steps output a Variant Call Format
(VCF) file. What's the difference? Look first
at the pileup file using zless:
zless -S GWD_FUT2_pileup.vcf.gz
The first lines in the file (the ones starting with a # character) are all metadata. They're
useful but let's skip them for now. The easiest way to do this is search for 'CHROM' which you can
do by pressing / (forward slash) followed by CHROM, and then press enter. You should see
something like this:
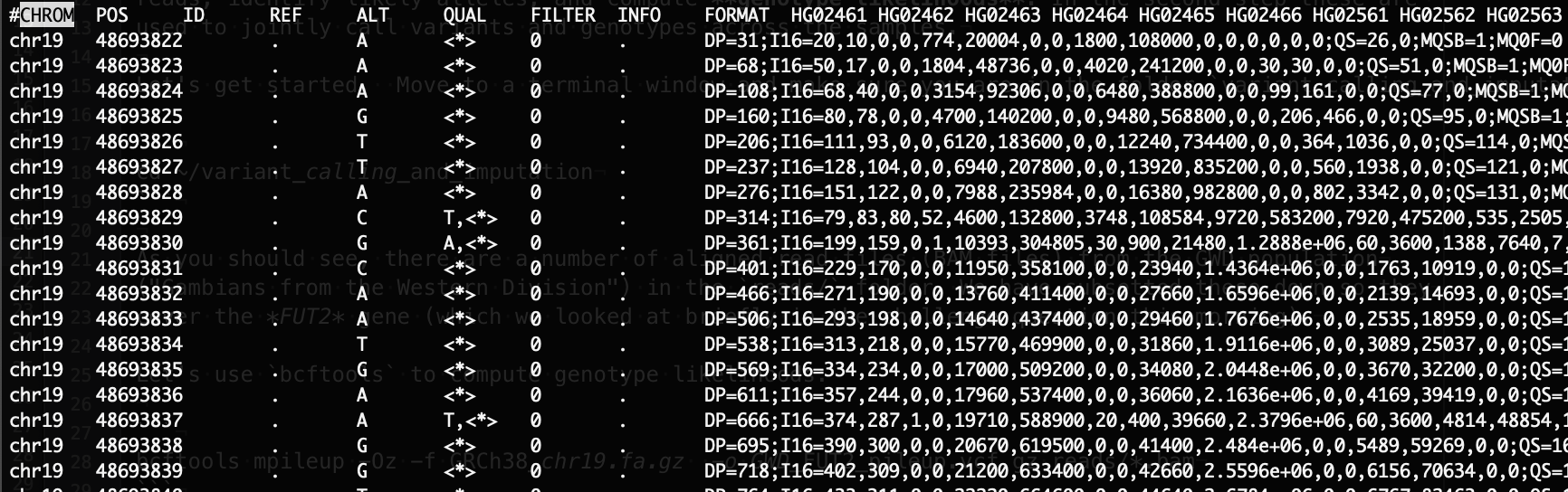
Feel free to scroll around a bit. A few things to note:
bcftools mpileuphas looked at every site in the region (from 48693822 to 48708100, as it happens) and assessed the evidence for variation at each one.The 4th and 5th columns tell you what alleles
mpileupthinks are present in the reads. The 4th column is the reference sequence base, and the 5th is are the other alleles seen (with<*>standing for 'anything else').The 8th column is called
INFOand contains a bunch of information about the variants across all samples. Exactly what is in there is described in the metadata, but for example you should be able to see values calledDP. This is the total depth of reads observed across all samples at the site. Other metrics similarly capture properties of the read pileup.The per-sample data itself starts way over in the 10th column. The values are PHRED-scaled genotype likelihoods for each possible genotype for the given alleles.
Note. If there are alleles listed, then there are possible genotypes (for a diploid human individual)
- that is, possible homozygous genotypes, and (read as " choose ", i.e. the number of ways of picking alleles from ) possible heterozygous genotypes. This is why some rows have many more values than others - more alleles were seen in the reads.
Note. The PHRED-scaled likelihood (PL) for a particular genotype is computed as
The simplest reasonable likelihood function in the bracket here would be something proportional to a binomial likelihood, but in practice bcftools uses a more complex likelihood that allows for error dependency between reads. (One motivation is that an error might occur as an alignment error or be sequence context-dependent, so it makes sense to allow for this.)
Example. For the first site and first sample, the PLs are 0, 3, 26. That translates to
likelihoods of about 1, 0.5, and 0.0025. pileup is not very certain about what this genotype is!
If you look at the sample using tview you'll see why:
samtools tview -p chr19:48693822 reads/HG02461.final_FUT2.bam
Only two reads cover this position in this sample - they're both A bases,
but more reads would be needed to be really confident this is a homozygous A.
Inspecting the calls output
Now look at the calls file:
zless -S GWD_FUT2_calls.vcf.gz
Again skip past the metadata by typing /CHROM and pressing ENTER. You should see something like this:
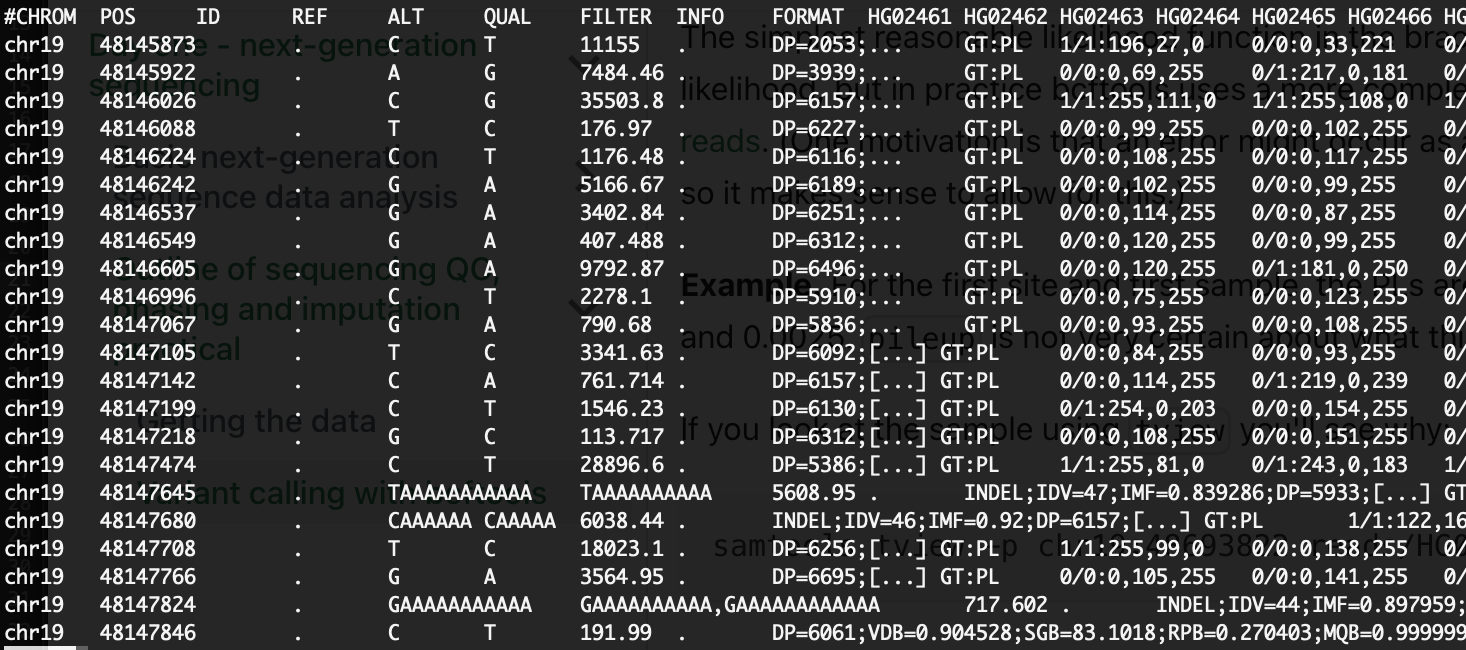
(NB. I've shrunk the INFO data in the above image to make it easier to see - your file will no doubt have a bunch of stuff in there).
A few points to note:
- The file only has a subset of sites in it - those where
bcftoolsthinks there is genuine variation. - The
<*>alleles are gone. They were just there to enable the calling step to sum evidence across samples. - The PLs are still there - but now they are joined by genotype calls in the
GTfield.
Scroll down the file a bit. Can you find a multi-allelic variant? (i.e. several alleles in column 5?) What about a multi-allelic SNP (rather than an indel?)
Interpreting the GT field
It's pretty simple. A 0 corresponds to the reference allele, a 1 corresponds to the first
alternative allele, and so on. For a diploid sample the genotypes are of the form 0/0 (homozygous
reference), 0/1 (heterozygous for the first alternative allele) 1/1 homozygous for the first
alternative allele, and so on.
By and large these genotypes correspond to the maximum likelihood genotype (as you can tell by comparing to the PL fields... remember we're looking for the smallest PL because of the PHRED scaling.) However, if there aren't many reads the genotype may also be influenced by the prior.
An aside on genotype posteriors.
In the lecture you will have seen that the calling works in a Bayesian way, by computing a
posterior probability of each genotype. But bcftools doesn't output that posterior by default.
To get it, you can change the calling command to add the -a GP option:
bcftools call -m -v -Oz -a GP -o GWD_FUT2_calls_with_GP.vcf.gz GWD_FUT2_pileup.vcf.gz
If you look at this new output file it has, in addition to genotype (GT) and PHRED-scaled
genotype likelihood (PL), a new field called GP which are the genotype posteriors. They look
like this:
#CHROM POS ID REF ALT QUAL FILTER FORMAT HG02461 HG02462 HG02463 HG02464 HG02465 HG02466 HG02561 HG02562 HG02563 HG02568 HG02569 HG02570 HG02571 HG02572 HG02573 HG02574 HG02575 HG02582 HG02583 HG02584 HG02585 HG02586 HG02588 HG02589 HG02590 HG02594 HG02595...
chr19 48693829 . C T 3357.45 . GT:PL:GP 0/0:0,9,75:0.810234,0.189766,2.21701e-08 1/1:85,9,0:2.87613e-09,0.213018,0.786982 0/1:21,0,51:0.00425148,0.995745,3.67872e-06 ./.:0,0,0:0,0,0 0/1:24,0,24:0.00213138,0.996024,0.00184...
chr19 48693956 . A G 6806.98 . GT:PL:GP 0/0:0,99,255:1,3.1458e-11,4.93632e-28 0/0:0,105,255:1,7.9019e-12,4.93632e-28 0/0:0,111,255:1,1.98487e-12,4.93632e-28 0/0:0,78,255:1,3.96033e-09,4.93632e-28 0/0:0,69,255:1,3.1458e-08,4.93632e-28...
chr19 48693992 . G A 1802.53 . GT:PL:GP 0/0:0,96,255:1,1.43086e-11,2.56527e-29 0/0:0,105,255:1,1.80134e-12,2.56527e-29 0/0:0,90,255:1,5.69634e-11,2.56527e-29 0/0:0,105,255:1,1.80134e-12,2.56527e-29 0/0:0,81,255:1,4.52477e-10,2.56527e-29...
chr19 48694122 . C G 125.986 . GT:PL:GP 0/0:0,144,255:1,2.25005e-17,2.52536e-31 0/0:0,114,255:1,2.25005e-14,2.52536e-31 0/0:0,102,255:1,3.56609e-13,2.52536e-31 0/0:0,111,255:1,4.48944e-14,2.52536e-31 0/0:0,93,255:1,2.83264e-12,2.52536e-31...
chr19 48694244 . GTTTTTTTTTTTTTTTTTTTTTT GTTTTTTTTTTTTTTTTTTTTT,GTTTTTTTTTTTTTTTTTTTTTTT 3261.26 . GT:PL:GP 0/1:31,0,37,52,46,77:0.00179946,0.998173,2.19386e-05,3.14376e-06,2.75729e-06,5.46634e-10 0/2:54,84,91,0,42,27:1.8098e-05,7.97434e-09,1.7...
chr19 48694267 . G T 186.473 . GT:PL:GP 0/0:0,42,255:0.999989,1.09366e-05,2.37526e-28 0/0:0,28,255:0.999725,0.000274643,2.37463e-28 0/0:0,30,248:0.999827,0.000173306,1.19026e-27 0/0:0,38,217:0.999973,2.74711e-05,1.49866e-24 0/0:0,8...
chr19 48694335 . G A 1408.47 . GT:PL:GP 0/0:0,81,255:1,4.55947e-10,2.60477e-29 0/0:0,84,255:1,2.28515e-10,2.60477e-29 0/0:0,58,255:1,9.09735e-08,2.60477e-29 0/0:0,75,255:1,1.81516e-09,2.60477e-29 0/0:0,93,255:1,2.87683e-11,2.60477e-29...
chr19 48694424 . T A 11282.2 . GT:PL:GP 0/0:0,102,255:1,3.49087e-11,2.41996e-27 1/1:255,90,0:4.13231e-25,7.2298e-09,1 0/1:202,0,151:1.14042e-20,1,1.09869e-16 0/0:0,108,255:1,8.76867e-12,2.41996e-27 0/1:200,0,202:1.80745e-20,1,8.72717e-22...
chr19 48694447 . G A 3214.56 . GT:PL:GP 0/1:180,0,241:8.19006e-18,1,2.42467e-26 0/0:0,105,255:1,3.86112e-12,1.1786e-28 0/1:161,0,240:6.50559e-16,1,3.05248e-26 0/0:0,108,255:1,1.93514e-12,1.1786e-28 0/0:0,81,255:1,9.69869e-10,1.1786e-28...
chr19 48694448 . G A 349.569 . GT:PL:GP 0/0:0,114,255:1,4.83668e-14,1.1669e-30 0/0:0,105,255:1,3.84191e-13,1.1669e-30 0/0:0,105,255:1,3.84191e-13,1.1669e-30 0/0:0,108,255:1,1.92552e-13,1.1669e-30 0/0:0,78,255:1,1.92552e-10,1.1669e-30...
chr19 48694648 . G A 360.549 . GT:PL:GP 0/0:0,105,255:1,4.222e-13,1.40921e-30 0/0:0,87,255:1,2.6639e-11,1.40921e-30 0/0:0,87,255:1,2.6639e-11,1.40921e-30 0/1:196,0,199:1.8814e-18,1,4.20202e-23 0/0:0,78,255:1,2.11601e-10,1.40921e-30...
chr19 48694728 . C A 3031.84 . GT:PL:GP 0/0:0,99,255:1,1.31458e-11,8.62013e-29 0/0:0,81,255:1,8.29443e-10,8.62013e-29 0/0:0,78,255:1,1.65496e-09,8.62013e-29 0/1:178,0,193:1.5178e-17,1,1.30836e-21 0/0:0,75,255:1,3.30207e-09,8.62013e-29...
For example, for the first sample at the first SNP, the posterior probabilities are , ,
and . So bcftools is reasonably confident that the genotype is homozygous
reference, but there's about a 20% chance it is heterozygous.
Note. The VCF spec lies about these fields. It says they are PHRED scaled, but they aren't - they are unscaled probabilities.
Questions
How many of your variants are SNPs? How many are INDELs? How many are multi-allelic and how many bi-allelic?
Next steps
The rest of this practical is about using the wealth of information bcftools has returned to turn
this initial set of variant calls into a robust set of phased genotypes. We'll then apply that to
impute a second set of samples. For this purpose we'll use a set of calls on a larger genomic
region, that you can find in the file calls/GWD_30X_calls.vcf.gz.
When you're ready go back to the practical.