Appendix 2: understanding sequence duplication levels
Duplicates arise naturally in sequencing from random fragments that just happen to have the same breakpoints. However, as explained a bit more on the paired-end sequencing theory page, they also arise artifically from amplification and other chemistry artifacts, so it makes sense to assess levels of duplication in the data. That's what the plot on the QC page does. For our sample, it says that most reads are not duplicated, but a few do appear twice or more.
Does that look sensible? Well, here are three considerations
Duplicates by chance!
If we sequence a genome to a high level of coverage we'll of course get some duplicates just by chance. To assess that, let's ignore the paired end-y-ness for a moment and imagine sequencing the malaria genome with 2 million single-end 100 base pair reads. We could simulate that by just choosing random read start points - here's a bit of R code that does that:
# make sure to run this in an R session - not in the terminal!
simulate_duplicates = function( genome_size, number_of_reads ) {
# Sample read start locations
read_locations = sample( 1:genome_size, size = number_of_reads, replace = T )
# tabulate where the reads hit and make a histogram.
reads_per_location = table( read_locations )
M = max( max( reads_per_location ), 10 )
# Make a histogram
h = hist(
reads_per_location,
breaks = 0.5 + 0:M,
plot = FALSE
)
# Count duplicates (one per each additional copy)
duplicates = sum( 1:(M-1) * h$counts[2:M] )
return( list(
duplicate_rate = duplicates / number_of_reads,
histogram = h
))
}
d = simulate_duplicates( 23E6, 2E6 )
This returns a duplication rate of about 4% - let's plot it:
plot(
1:10,
d$histogram$density * 100,
xlab = "Sequence duplication level",
ylab = "Proportion (%)",
type = 'l',
ylim = c(0, 100),
bty = 'n'
)
This shows:
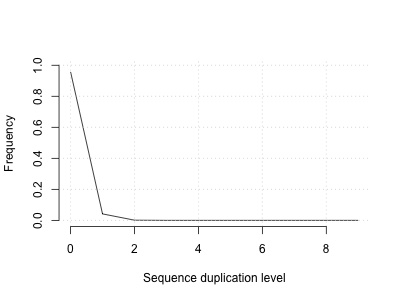
So what this suggests is that we ought to get some duplicates anyway - even if everything else was perfect. (In reality, sequence coverage is far from uniform so we could expect a higher number of duplicates)
Sequencing errors harm detection
FastQC's method of detecting duplicates relies on the first 50bp of duplicates being identical. However the first few bp also typically have higher error rates, which might hurt detection. So this might lead fastqc to underestimate the duplication rate.
Fastqc doesn't take into account read pairs
Another point to note is that fastqc does not take into account the read pairs in this analysis - it analyses each fastq file seperately. Artifically-generated duplicates generally duplicate the whole fragment, meaning that both read 1 and read 2 should be duplicated. So this might lead fastqc to overestimate the duplication rate.
Genomes contain true duplicates
It's worth noting also that genomes contain truly duplicated sequence. So some level of duplication may arise from reads that are not really read duplicates, but simply reads coming from duplicated sequence.
Conclusion
My hunch is that the 3% duplication rate output by fastqc above is probably an underestimate.
One way to find out is to try comparing the number above to the number of reads that are marked as duplicates in the alignment section. Those duplicates are computed based on the alignment positions of read 1 and read 2, and are probably more accurate.