A naive approach
Figuring out the proportion in genes should be pretty easy using the dplyr data verbs, right?. We could
- group genes by species and chromosome
- compute the total length by summing
- join to the
contigsdataset to compute total chromosome/region length - and then summarise.
Here's a function that does 1-3 above:
compute_lengths_per_chromosome = function(
data,
chromosomes = contigs
) {
(
data
# Group by species / chromosome
%>% group_by( dataset, seqid )
# Add up the gene lengths
%>% summarise(
number_of_genes = n(),
total_gene_length = sum( end - start + 1 )
)
# Add the chromosome lengths
%>% left_join(
chromosomes[, c( "dataset", "seqid", "attributes", "sequence_length" )],
by = c( "dataset", "seqid" )
)
)
}
The idea is that this has both a 'total gene length' (sum of lengths of genes) and the total sequence length (length of the chromosome or contig) in the same dataframe:
You can use it like this:
print( compute_lengths_per_chromosome( genes ) )
# A tibble: 5,469 × 6
# Groups: dataset [11]
dataset seqid number_of_genes total_gene_length attributes sequence_length
<chr> <chr> <int> <dbl> <chr> <dbl>
1 Acanthoch… MVNR… 56 1349511 Alias=orp… 2466117
2 Acanthoch… MVNR… 39 1029333 Alias=orp… 2338421
3 Acanthoch… MVNR… 56 1399228 Alias=orp… 2213793
4 Acanthoch… MVNR… 41 1113000 Alias=orp… 2013838
5 Acanthoch… MVNR… 45 1012078 Alias=orp… 1950346
6 Acanthoch… MVNR… 54 989013 Alias=orp… 2094391
7 Acanthoch… MVNR… 84 1074990 Alias=orp… 1729193
8 Acanthoch… MVNR… 72 1486459 Alias=orp… 2368326
9 Acanthoch… MVNR… 50 1237060 Alias=orp… 2396890
10 Acanthoch… MVNR… 39 643138 Alias=orp… 1656807
And all we have to do is step 4 - sum them up across chromosomes:
naive_proportions = (
compute_lengths_per_chromosome( genes )
%>% group_by( dataset )
%>% summarise(
total_gene_length = sum( total_gene_length ),
total_genome_length = sum( sequence_length )
)
%>% mutate(
proportion_in_genes = total_gene_length / total_genome_length
)
)
print( naive_proportions )
# A tibble: 11 × 4
dataset total_gene_length total_genome_length proportion_in_genes
<chr> <dbl> <dbl> <dbl>
1 Acanthochromis_polyacanthus.ASM210954v1.110 422171067 830201259 0.509
2 Asparagus_officinalis.Aspof.V1.57 165883925 1015569172 0.163
3 Bufo_bufo-GCA_905171765.1-2022_05-genes 1290309576 5003028965 0.258
4 Camelus_dromedarius.CamDro2.110.chr 825762826 2052758708 0.402
5 Gallus_gallus.bGalGal1.mat.broiler.GRCg7b.110.chr 583352821 1041139641 0.560
6 Homo_sapiens.GRCh38.110.chr 1379802830 3088286401 0.447
7 Mus_musculus.GRCm39.110.chr 1070748851 2723431143 0.393
8 Pan_troglodytes.Pan_tro_3.0.110.chr 1110722691 2967125077 0.374
9 Plasmodium_falciparum.ASM276v2.57 13776689 23292622 0.591
10 Plasmodium_knowlesi.ASM635v1.57 12970692 23462187 0.553
11 Plasmodium_vivax.ASM241v2.57 14052272 23768694 0.591
To figure out how much of the genome is covered by genes, or by exons, we face a problem.
In principle we could just add together the gene lengths.
(
ggplot( data = proportions )
+ geom_col(
aes(
x = proportion_in_genes,
y = dataset
)
)
+ ylab( "Species" )
+ xlab( "Proportion of genome in genes" )
+ theme_minimal(16)
)
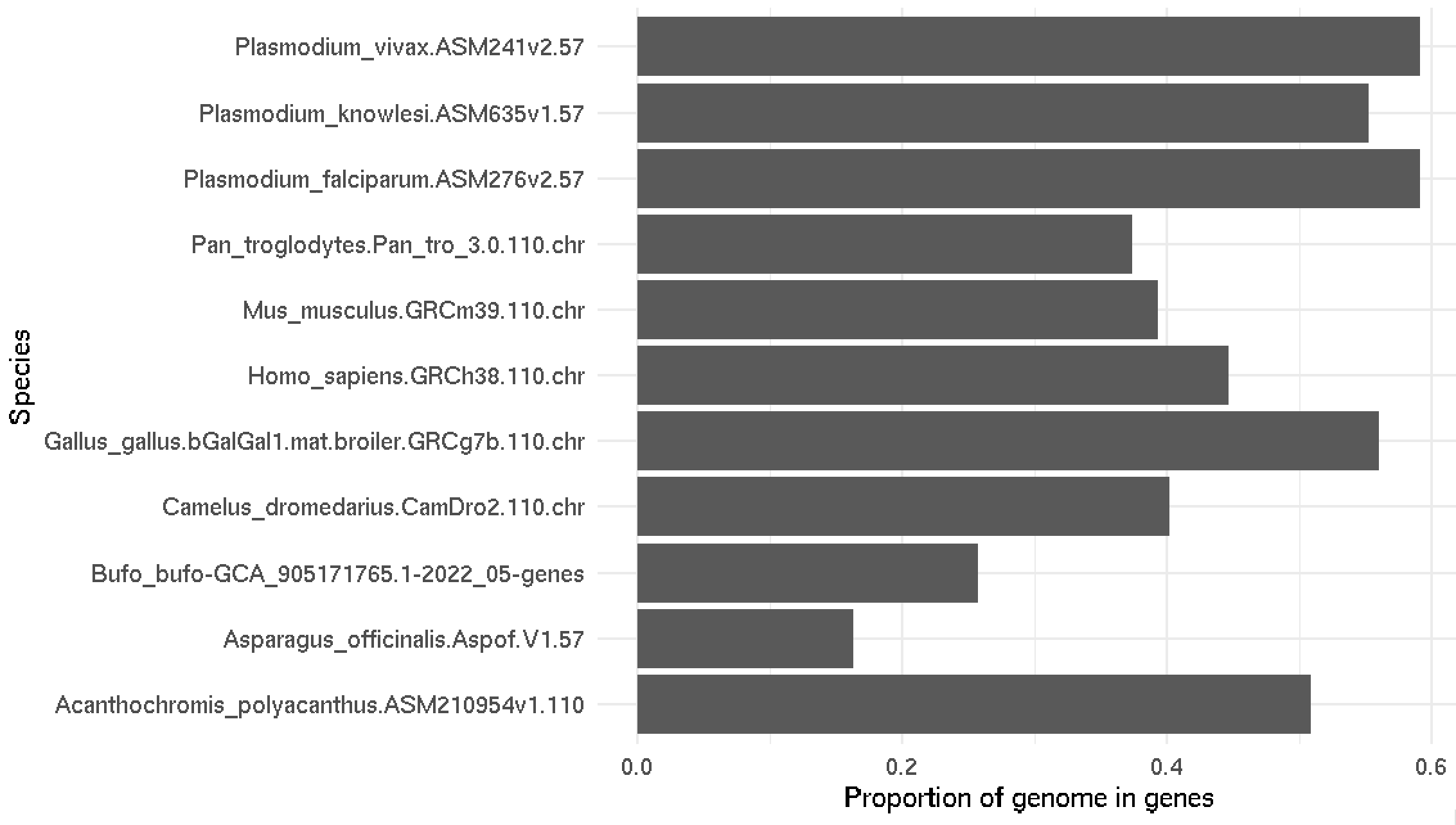
Pretty cool huh! Most species have 40-60% of the genome in genes. Is this right?
However
Unfortunately this calculation is (slightly) wrong. Why?
To get it right, we need to do some more coding...